
A surviving beta cell subpopulation enriched in patients with T1D - Max Spurrell MD/PhD candidate- Yale University
Pregame: Ahead of the June 16 talk

TheSugarScience T1D Th1nk Tank
📅 Registration
Date: Tuesday, June 16, 2026 · 12:00 PM Eastern Format: Free virtual seminar for the global T1D research community — clinicians and scientists are welcome.
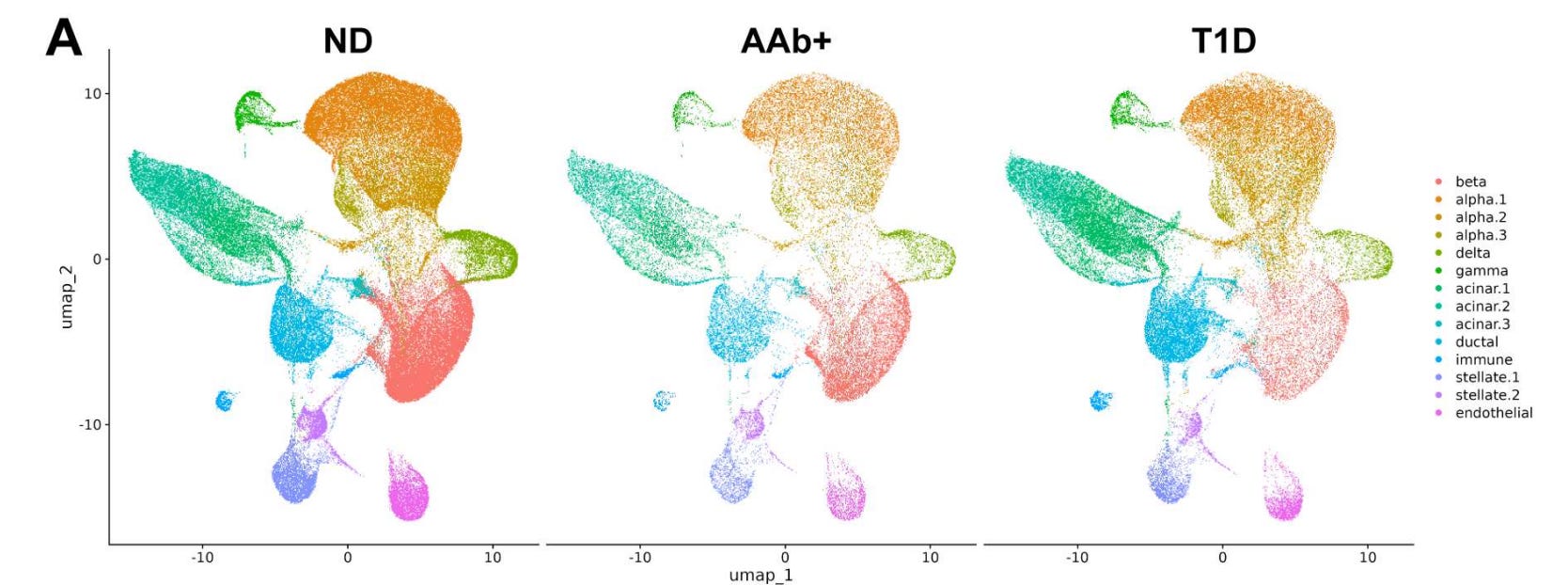
Figure adapted from DOI: 10.64898/2026.05.15.725449
👤 About the Speaker
Maxwell Spurrell is an MD/PhD student in the Department of Immunobiology at Yale School of Medicine, co-mentored by Drs. Kevan Herold and John Tsang. He grew up in Providence, RI, and completed his undergraduate studies at Brown University before joining the laboratory of Dr. Nir Hacohen at Massachusetts General Hospital, where he studied how the spatial organization of the tumor microenvironment in human lung cancer shapes responses to immunotherapy. That foundation in the computational and spatial biology of immune-tissue interactions now informs his graduate work at Yale, where he has redirected his attention from cancer immunology to the immunology of the pancreatic islet — specifically, to the question of why a small number of beta cells survive for years and even decades in a tissue under active immune assault.
His mentors situate him at a productive intersection. Kevan Herold is one of the preeminent figures in T1D immunology and beta cell biology, known internationally for his foundational work developing and clinically validating teplizumab — the first FDA-approved drug to delay T1D onset. John Tsang is a systems immunologist with deep expertise in single-cell multi-omics and gene regulatory network inference, and directs Yale’s Center for Systems and Engineering Immunology. Spurrell has combined their approaches to ask a question that requires both: what is the transcriptional state of the beta cells that are still alive in a patient who has had T1D for years — and what is it about them that made them survive?
01 · The Paper
“A Surviving Beta Cell Subpopulation Enriched in Patients with T1D” Maxwell Spurrell, John S. Tsang & Kevan C. Herold · bioRxiv, May 2026 DOI: 10.64898/2026.05.15.725449 · https://www.biorxiv.org/content/10.64898/2026.05.15.725449v2
The canonical story of T1D is a story of destruction: autoimmune T cells systematically eliminate the insulin-producing beta cells until essentially none remain, insulin production ceases, and the patient becomes dependent on exogenous insulin for life. That story is mostly right — but it is not entirely right, and the exceptions are biologically significant. A meaningful fraction of T1D patients, especially those diagnosed in adulthood, retain measurable residual insulin secretion for years after onset, as detected by circulating C-peptide. And at the histological level, cadaveric studies consistently find residual beta cells — scattered insulin-positive cells — in islets from donors who had T1D for decades. Something protects these cells. The question is what.
Studying these surviving cells has been technically formidable. They are rare, embedded in tissues that can only be accessed after death, and difficult to distinguish from neighboring cells without high-resolution single-cell methods. Prior work in mouse models — notably the Rui et al. 2017 paper identifying a “Btm” (bottom) subpopulation of beta cells in NOD mice that preferentially survived immune attack and expressed lower autoantigen levels alongside higher PD-L1 — established that immune-resistant beta cell subpopulations exist in principle. But whether an analogous state exists in human T1D, and what its molecular characteristics are, has remained unresolved.
Spurrell and colleagues address this question using the Human Pancreas Analysis Program (HPAP) single-cell RNA sequencing dataset — the largest available scRNA-seq resource for human pancreatic islets, drawn from cadaveric donors with T1D, autoantibody-positive donors at risk for T1D, and non-diabetic controls. Rather than relying on conventional clustering of gene expression alone, they apply a gene regulatory network (GRN) inference-based approach, using the SCENIC framework to infer transcription factor activity patterns and cluster beta cells based on the regulatory programs active within them. This methodological choice turns out to be decisive — it reveals a beta cell subtype that conventional approaches would have partially obscured.
The key findings:
A novel beta cell cluster (C3) is selectively enriched in T1D donors. GRN-based clustering identified a discrete beta cell subpopulation — Cluster 3 (C3) — that is markedly overrepresented in donors with established T1D compared to autoantibody-positive pre-diabetic donors and non-diabetic controls. This cluster represents the surviving beta cell population in human T1D and is defined not by gene expression alone but by a distinctive pattern of transcription factor activity.
IRF1 is the dominant transcription factor defining the C3 state. The C3 cluster is characterized by elevated activity of several transcription factors with established roles in beta cell survival, with IRF1 — interferon regulatory factor 1 — emerging as the most prominent. IRF1 has been previously shown to modulate beta cell survival in allograft models and to suppress inflammatory chemokine production; its enrichment here places it at the center of a regulatory program that may distinguish surviving from eliminated beta cells.
C3 beta cells upregulate immunomodulatory genes and downregulate autoantigens. The C3 population shows increased expression of SOCS1, SOCS3, and HLA-E — genes associated with suppression of immune signaling and resistance to NK cell- and T cell-mediated killing — alongside decreased expression of known T1D autoantigens and secretory pathway genes. This combination is consistent with a dedifferentiation program that trades functional identity for immune invisibility.
Inflammatory cytokines drive the C3 transcriptional state. Reanalysis of published data from primary human beta cells stimulated with inflammatory cytokines in vitro showed that cytokine exposure induces a transcriptional program that closely resembles the C3 state — placing cytokine signaling, not some pre-existing intrinsic property of a rare beta cell subtype, as the likely environmental driver of this phenotype.
A parallel program is active in a subset of alpha cells. The C3-like transcriptional program is also detected in a fraction of alpha cells from T1D donors — a finding consistent with cell-extrinsic cytokine signaling acting on all endocrine cells in the inflamed islet environment, not just beta cells, and suggesting that the resilience phenotype may reflect a general response to chronic interferon exposure rather than a beta cell-specific fate.
Tags: beta cell heterogeneity · scRNA-seq · HPAP · GRN inference · SCENIC · IRF1 · SOCS1 · HLA-E · dedifferentiation · immune evasion · cytokines · T1D · surviving beta cells · nPOD · teplizumab · C-peptide · human pancreas · alpha cells
02 · Why This Matters
For scientists: The application of gene regulatory network inference (SCENIC) to identify beta cell subpopulations rather than clustering on raw gene expression is a methodological contribution that the field should notice. Conventional clustering approaches are designed to find groups defined by what genes are expressed; GRN-based approaches find groups defined by what regulatory circuits are active. These are not the same question, and for a population as transcriptionally stressed and partially dedifferentiated as surviving T1D beta cells, the regulatory program may be more informative and more stable than the expression of any individual gene. The identification of IRF1 as a central node in the survival program opens a direct mechanistic hypothesis: that IRF1 activity in beta cells, by suppressing autoantigen expression, upregulating checkpoint ligands, and dampening inflammatory chemokine production, generates a state of partial immune tolerance within the inflamed islet microenvironment. This is a testable model with multiple molecular handles.
The parallel finding in alpha cells is also scientifically significant. If the same transcriptional program is active in a fraction of alpha cells from T1D donors, it suggests that the surviving-cell phenotype is not a beta cell-intrinsic fate program but a tissue-wide response to chronic cytokine exposure. This reframes the question: rather than asking what is special about the surviving beta cells, we should be asking what is special about the cytokine environment they experienced — and whether that environment can be recreated, modified, or exploited therapeutically.
For clinicians: The survival program identified here is not an idle biological curiosity — it is a potential therapeutic target. If surviving beta cells are maintained in part by IRF1-driven upregulation of SOCS1/3 and HLA-E, and if inflammatory cytokines can drive naive beta cells into this state, then there may be a window in early or pre-clinical T1D during which cytokine signaling could be modulated to tip more beta cells into a survival-competent phenotype before they are eliminated. This connects directly to the clinical program of immunotherapy for T1D — particularly teplizumab, which Herold’s lab developed and which is now FDA-approved. Understanding what allows residual beta cells to survive immune attack is mechanistically relevant to understanding why teplizumab works in some patients and not others, and to identifying new combination strategies that could augment its effect.
The broader picture: The question of what allows some beta cells to survive T1D for decades has been asked for a long time, but it has largely been unanswerable because the tools to study rare human cells in their native tissue context at single-cell resolution did not exist at scale. The HPAP dataset, combined with methods like SCENIC that can extract regulatory logic from sparse scRNA-seq data, now makes this question tractable in human tissue. This paper is one of the first to fully exploit that combination to define, at the transcription factor level, what surviving human T1D beta cells actually are. That definition — IRF1-active, SOCS-expressing, autoantigen-low, dedifferentiated — is specific enough to generate hypotheses, design experiments, and think about therapeutic leverage. That is a meaningful step forward.
03 · Four Questions We Will Ask the Speaker
Q1. Your GRN-based clustering approach revealed the C3 population where conventional expression-based clustering did not — or at least not as cleanly. Can you describe intuitively what SCENIC is detecting that standard Seurat-style clustering misses, and how confident you are that C3 is a discrete stable cell state rather than a transitional point on a continuous stress trajectory?
Q2. IRF1 sits at the center of the C3 regulatory program, and you show cytokine stimulation can drive cells into a C3-like state in vitro. Is there a mechanistic hypothesis for how IRF1 specifically protects against immune killing — is it primarily through autoantigen downregulation, through SOCS-mediated dampening of cytokine signaling, through HLA-E-mediated inhibition of NK cells, or do you think all of these are required together?
Q3. The C3-like program is also found in a subset of alpha cells from T1D donors — cells that are not being targeted by autoimmunity. Does the presence of the same transcriptional state in alpha cells help you disentangle whether this is a cell-autonomous survival response or purely a product of the local cytokine environment? And does it tell you anything about whether the C3 state is protective or simply a bystander marker of cytokine exposure?
Q4. The translational implication you gesture toward is that the C3 program might be a target for T1D prevention or reversal. If IRF1 activity drives survival, would you want to induce this state in more beta cells early in disease — and if so, how would you do that without compromising insulin secretory function, given the associated dedifferentiation signature?
04 · Four Key Associated Papers
Rui J, Deng S, Arazi A, Bhatt AS, Liu Z & Herold KC (2017) β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice · Cell Metabolism, 25(3):727–738 · READ HERE · The foundational mouse study from the Herold lab identifying a discrete subpopulation of beta cells (”Btm” cells) that survive immune attack in NOD mice, characterized by lower autoantigen expression and higher PD-L1 — the direct conceptual and experimental predecessor of today’s human study. Today’s paper can be read as the human translational answer to the question Rui et al. first posed in mice.
Fasolino M, Schwartz GW, Patil AR, Mongia A et al. (2022) Single-cell multi-omics analysis of human pancreatic islets reveals novel cellular states in type 1 diabetes · Nature Metabolism, 4:284–299 · READ HERE · The HPAP-generated single-cell atlas of 24 T1D, autoantibody-positive, and non-diabetic human donor islets using scRNA-seq, CyTOF, and imaging mass cytometry — the foundational dataset that Spurrell and colleagues directly build on and reanalyze to identify the surviving beta cell subpopulation.
Elgamal R et al. & Kaestner KH (2023) Computational workflow and interactive analysis of single-cell expression profiling of islets generated by the Human Pancreas Analysis Program · Nature Metabolism, 5:2025–2035 · READ HERE · The expanded HPAP scRNA-seq resource paper covering 258,000+ cells from 67 donors across non-diabetic, autoantibody-positive, T1D, and T2D categories — the updated version of the dataset providing the beta cell observations analyzed in today’s paper, and an essential reference for understanding the scope and structure of the data.
Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W et al. (2013) Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial · Diabetes, 62(11):3766–3774 · READ HERE · Herold’s pivotal clinical trial demonstrating that teplizumab preserves residual beta cell function (measured by C-peptide) in new-onset T1D — the clinical evidence that residual beta cells are functionally relevant and worth preserving, and the direct clinical-translational context that makes understanding why some beta cells survive immunologically important.
05 · Four Videos to Watch First
▶ Ask the Expert: Kevan Herold, MD, with Matthias Von Herrath, MD — Yale & UCSD TheSugarScience Ask the Expert featuring today's speaker's mentor Dr. Herold in conversation with Dr. Von Herrath, discussing teplizumab and opportunities for immunotherapy in T1D. Watch this first to understand the clinical context — the teplizumab program and the preservation of residual beta cell function — that directly motivates the question Max Spurrell is asking.
▶ Ask the Expert: Ruth Elgamal, PhD Candidate, UCSD — Integrated Pancreatic Islet Reference Map TheSugarScience Ask the Expert with Ruth Elgamal from the Kyle Gaulton lab, discussing the integrated HPAP islet reference map. Elgamal is a co-author on the HPAP dataset paper that Spurrell directly reanalyzes — this is the best introduction to the resource, its structure, and how to navigate it.
▶ Ask the Expert: Kyle Gaulton, PhD, UCSD — T1D Risk, Genetics, and Single-Cell Epigenetics TheSugarScience Ask the Expert with Dr. Kyle Gaulton, one of the leaders of the HPAP analytical effort, discussing how single-cell approaches to human islet biology are changing our understanding of T1D genetic risk. Essential context for understanding the single-cell technology landscape that makes studies like Spurrell’s possible.
▶ Ask the Expert: Braulio Marfil-Garza, PhD, with James Shapiro, MD — Islet Transplantation in T1D TheSugarScience Ask the Expert on 20-year islet transplantation outcomes. The relevance here is as a frame for the translational stakes: if surviving beta cells in T1D patients can be understood and potentially protected or expanded, the implications extend not just to immunotherapy but to the broader question of how much functional beta cell mass can be preserved or restored — a question central to both immunotherapy and cell replacement strategies.
TheSugarScience · Expediting a cure for T1D by curating the scientific conversation · thesugarscience.org